gMCSpy.calculateGMCS module
- buildGMatrix(cobraModel: Model, maxKOLength: int = 1000000.0, name: str | None = None, isoformSeparator: str | None = None, verbose: int = 0, **kwargs)[source]
Build the G matrix, G dict and the relationships between perturbations
The G matrix is a matrix with the perturbations of the genes in the model, that render a reaction infeasible. Each row of the matrix corresponds to a perturbation, and each column corresponds to a reaction. This matrix is used to calculate the genetic minimal cut sets of the model.
- Parameters:
cobraModel – The cobra model that represents the metabolism of an organism.
name – The name of the model. If None, the id of the model will be used.
separateIsoform – If not None, the isoforms of the genes will merge into one gene.
verbose – The level of verbosity of the function.
- Returns:
A List with the G matrix, the G dictionary and the relationships between perturbations.
- buildReactionFromBranch(expr: BoolOp, model: Model, level: int = 0, parentMetabolite: Metabolite | None = None) Model[source]
Recursive function to generate metabolic model from GPR.
Each GPR can be expressed as a metabolic model. To that end, a boolean tree can be build, where a branch can be either an AND, an OR or a gene. We move through the tree, and recursively call this function on each node. Until all genes are explored (end nodes). As we move through the tree, we add reactions to the model. The reactions are named with a unique identifier, to avoid name conflicts. An example building a metabolic model from a GPR is shown below.
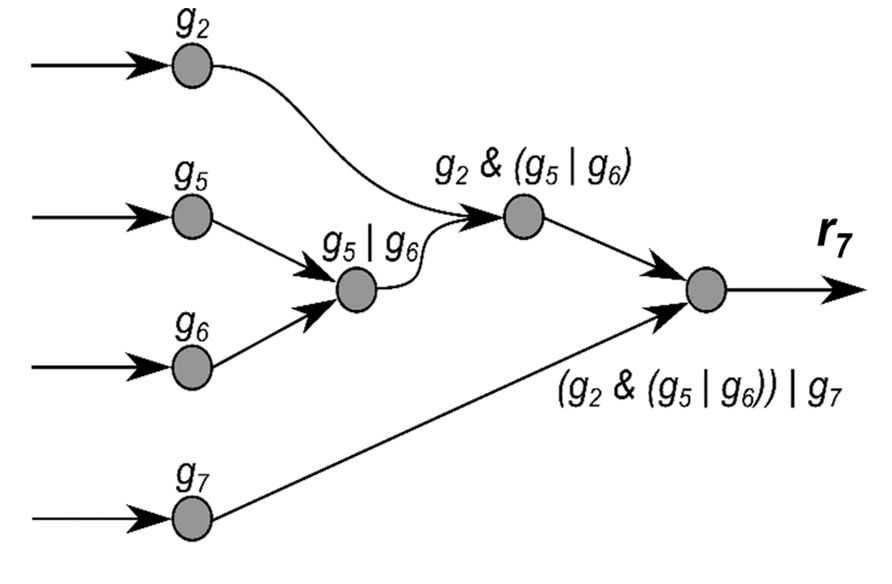
Figure 1. Example of a binary tree.
In the example above, the GPR is (g2 and (g5 or g6)) or g7. The function will be called recursively on each node.
- Parameters:
expr – A BoolOp object from the ast module to be explored. i.e. a tree. (ast.BoolOp)
model – A cobra model to build add the tree.
level – The level of the tree, i.e. the depth of the current node. (int)
parentMetabolite – The metabolite that is the parent of the current node. (cobra.core.Metabolite)
- Returns:
model: A cobra model with the tree added as reactions, genes have become metabolites, and the objective function set to the root node, the reaction of interest.
- buildRxnGeneMat(GPRs: List, reactions: List, genes: List)[source]
Build a reaction-gene matrix from a list of GPRs.
The binary matrix is build from each reaction GPR, each row corresponds to a reaction and each column corresponds to a gene. The matrix is sparse and is stored in the CSR format.
- Parameters:
GPRs – List of GPRs from a cobra model.
reactions – List of reactions from a cobra model.
genes – List of genes from a cobra model.
- Returns:
rxnGeneMatrix: A sparse matrix with the number of reactions in rows and the number of genes in columns. (scipy.sparse.csr_matrix)
summary: A dictionary with the number of reactions classified by the number of genes in their GPR. (dict)
- calculateGeneMCS(cobraModel, **kwargs)[source]
Calculate the genetic minimal cut sets of a model Calculates the gMCS of a metabolic network using a mixed integer linear programming (MILP) formulation. Among all genes or with a given subset of genes, the function finds the minimal set of genes to stop biomass production.
- Required arguments:
cobraModel: A metabolic model in cobrapy format. See cobra models.
- Optional arguments:
Solver: Solver to be used. Current compatibility includes cplex, gurobi and SCIP. Default value is gurobi.
maxKOLength : Maximum length of gMCS to be found. (i.e. number of genes in the gMCS). Default value is 3
maxNumberGMCS : Maximum number of gMCS to be found. Default value is 1e6
verbose: If 0, the solver will not print any information. Verbose = 1, log will be printed into the console. Verbose = 2, log will be printed into the console and a log file will be created. Default value is 0.
earlyStop : If True if optimality returned by the solver the solver will stop without running a second time. Default value is True.
removeRelated: If True, the solver will remove the gMCS that are a subset of another gMCS from the solution space. Default value is True.
targetKOs: List of genes that you want to be part of the gMCS solutions. The list is expected to be of gene names. Default value is an empty list, which means that all genes will be considered. The gMCS solutions will contain at least one of the genes in the list.
geneSubset: List of genes to be considered in the gMCS problem. The list is expected to be of gene names. Default value is an empty list, which means that all genes will be considered. This is useful when you want to reduce the search space of the gMCS problem. e.g. geneSubset = [“gene1”, “gene2”, “gene3”] will only consider genes gene1, gene2 and gene3 as possible intervention points. Only [“gene1”], [“gene2”], [“gene3”], [“gene1”, “gene2”], [“gene1”, “gene3”], [“gene2”, “gene3”] and [“gene1”, “gene2”, “gene3”] could be solutions. This not compatible with targetKOs.
isNutrient: If True, the solver will consider the exchange reactions as nutrients, assigning artificial genes to exchange reactions to evaluate what metabolites and genes are essential. Default value is False.
exchangeReactions: List of exchange reactions to be considered as nutrients. The list is expected to be of reaction names. Default value is None.
saveGMatrix: If True, the solver will save the G matrix in a csv file. Default value is False.
checkSolutions: If True, the solver will check if the solutions are valid. Default value is False.
saveSolutions: If True, the solver will save the solutions in a csv file. Default value is True.
timelimit: Maximum time allowed for the solver to find a solution. Default value is 1e75.
forceLength: If True, the solver will find MCS of length 1, 2, 3, …, MAX_LENGTH_MCS. If False, the solver will find MCS of length MAX_LENGTH_MCS. Default value is True for cplex and false for gurobi.
numWorkers: Number of workers to be used for parallel processing. Default value is 0. The solvers cplex and gurobi already use parallel processing.
isoformSeparator: If not None, the isoforms of the genes will merge into one gene. Default value is None.
- calculateMCSForGPRs(briefing: dict, isoformSeparator: str, verbose: int = 0, **kwargs)[source]
Calculate the Minimal Cut Sets for GPRs with both AND and OR relationships
Given a complex GPR, we parse de GPR to a cobra model and calculate the MCSs for the model.
- Parameters:
briefing – The dictionary with the information of the GPRs.
verbose – The verbosity level.
- Returns:
A List with the List of GPRs and a List with the MCSs.
- createSparseMatrix(gMatrixDict, modelReactions)[source]
Build the G matrix
Create sparse matrix from the dictionary with the perturbations.
- Parameters:
gMatrixDict – The dictionary with the perturbations.
modelReactions – The reactions of the model.
- Returns:
The G matrix.
- parseGPRToModel(stringGPR: str, reactionName, isoformSeparator)[source]
Parse a GPR string to a cobra model
The model is used to calculate the MCS to calculate the combinations of genes that are required for the reaction.
- Parameters:
stringGPR – The GPR string to parse. (e.g. “gen1 and gen2 or gen3”)
reactionName – The name of the reaction to parse. Just to identify the model.
- Returns:
treeGenes - A list of genes that participate in the GPR.
model - A cobra model with the GPR parsed to a metabolic model.
Find the rows that are related to each other
If a row is a subset of another row, then that means that when the perturbation of the superset is activated, the subset is also affected. i. e. gene A and gene B inactivates reaction1, and gene A inactivates reaction2. Knocking out gene A and gene B will affect both reactions.
- Parameters:
gMatrixDict – The dictionary with the perturbations.
- Returns:
A list with the rows that are related to each other.
- simplifyGMatrix(gMatrixDict: defaultdict, __addToDict) defaultdict[source]
Simplify the G matrix
Analyze the G matrix to find the rows that are related to each other. If a common gene is more perturbations, then the perturbations are related to each other. And should be an individual perturbation.
- Parameters:
gMatrixDict – The dictionary with the perturbations.
- Returns:
The simplified G matrix.